Downloadable files
The ScPCA Portal download packages include gene expression data, a QC report, and associated metadata for each processed sample.
Gene expression data is available as either SingleCellExperiment objects (.rds files) or AnnData objects (.h5ad files).
All downloaded files are delivered as a zip file.
When you uncompress the zip file, the root directory name of your download will include the date you accessed the data on the ScPCA Portal.
We recommend you record this date in case there are future updates to the Portal that change the underlying data or if you need to cite the data in the future (see How to Cite for more information).
Please see our CHANGELOG for a summary of changes that impact downloads from the Portal.
Data can be downloaded by either downloading a single project, creating a custom dataset, or by choosing one of the Portal-wide download options.
For all data downloads, sample folders (indicated by the SCPCS prefix) contain the files for all libraries (SCPCL prefix) derived from that biological sample.
Most samples only have one library that has been sequenced.
For multiplexed sample libraries, the sample folder name will be an underscore-separated list of all samples found in the library files that the folder contains.
Note that multiplexed sample libraries are only available as SingleCellExperiment objects, and are not currently available as AnnData objects.
See the FAQ section about samples and libraries for more information.
The files shown below will be included with each library (example shown for a library with ID SCPCL000000):
An unfiltered counts file:
SCPCL000000_unfiltered.rdsorSCPCL000000_unfiltered_rna.h5ad,A filtered counts file:
SCPCL000000_filtered.rdsorSCPCL000000_filtered_rna.h5ad,A processed counts file:
SCPCL000000_processed.rdsorSCPCL000000_processed_rna.h5ad,A quality control report:
SCPCL000000_qc.html,A supplemental cell type report:
SCPCL000000_celltype-report.html
For more information on the contents of these files, see the sections on gene expression data, the QC report, and the cell type report.
Every download also includes sample and processing metadata for all libraries included in the download. For a full description of the metadata files, refer to the metadata section below.
Metadata-only downloads are also available, either by downloading the metadata for all samples in a single project using the Download Sample Metadata button or by downloading the metadata for all samples on the Portal.
Project downloads
Use the Download Now button next to the project title to instantly download gene expression data for all samples in a single project as a single zip file.
To download more than one project or combine samples across projects, see the section on downloading custom datasets.
For project downloads, data for all samples will be provided as either SingleCellExperiment objects (.rds files) or AnnData objects (.h5ad files).
Each zip file will be named with the project accession ID, the chosen data format (either single-cell-experiment or anndata), and the date you accessed the data on the ScPCA Portal.
If the project contains bulk RNA-seq data, a separate folder labeled with the
_bulksuffix containing two tab-separated value files,SCPCP000000_bulk_quant.tsvandSCPCP000000_bulk_metadata.tsv, will also be included in the project download. See the section on bulk RNA-seq for more information.If the project contains samples with a spatial transcriptomics library, the spatial data will be provided as a separate download. See the expected file structure and description of the Spatial transcriptomics output below.
If the project contains samples that have been multiplexed, the organization of the downloaded files will be slightly different than what is shown below. See the section describing multiplexed sample libraries for an overview of the expected download structure.
For more information on choosing a data format and modality, see the documentation on download options.
When downloading a project, you can choose to download data from all samples as individual files, or you can download a single file containing all samples merged into a single object. Below are examples of the expected folder structure when downloading a project with gene expression data from all samples stored in individual files.
Download folder structure for SingleCellExperiment project downloads:

Download folder structure for AnnData project downloads:

Download folder structure for AnnData project downloads with CITE-seq (ADT) data:

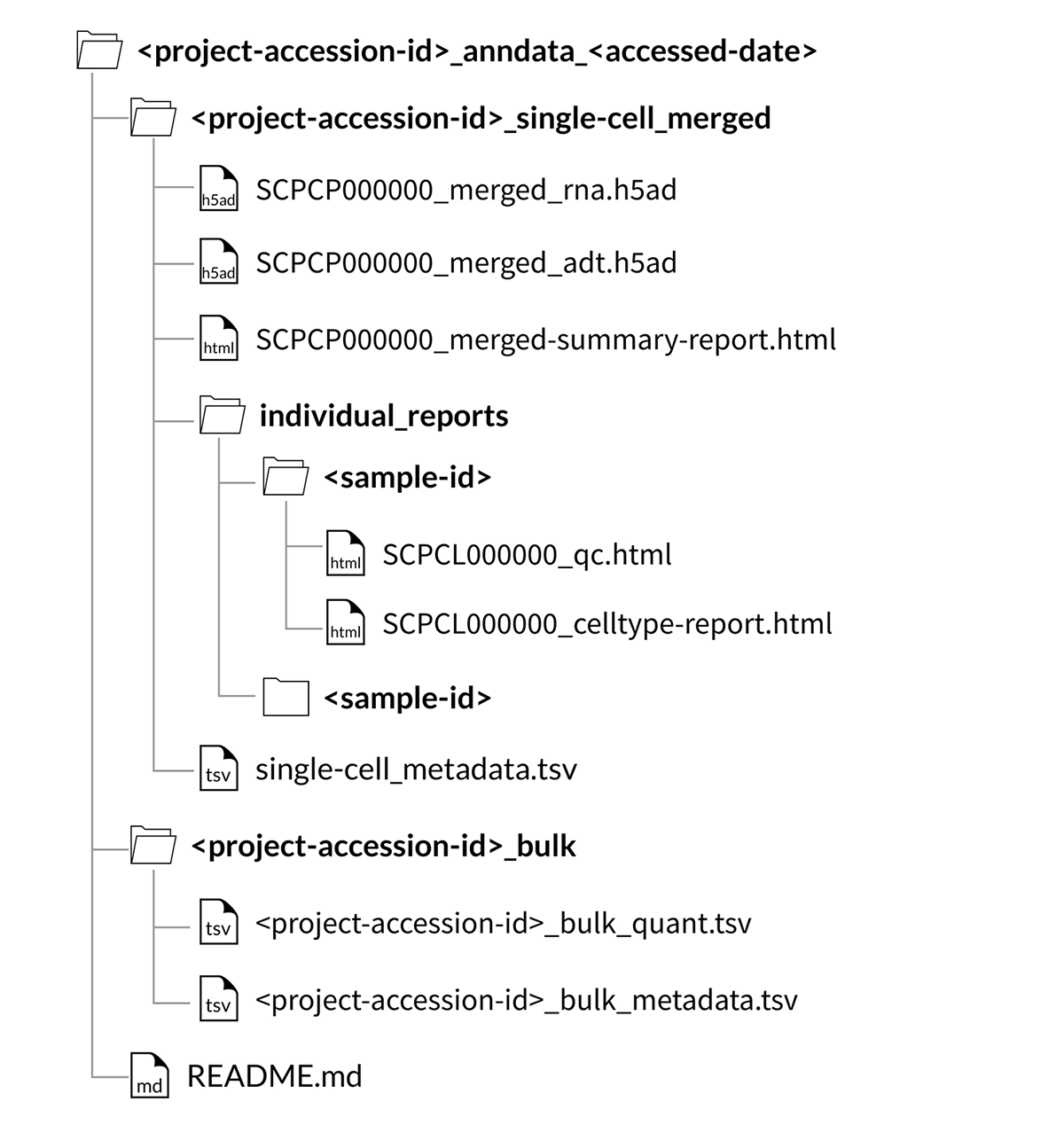
If downloading a project with samples that contain a CITE-seq library as an AnnData object (.h5ad file), the quantified CITE-seq expression data is included as a separate file with the suffix _adt.h5ad.
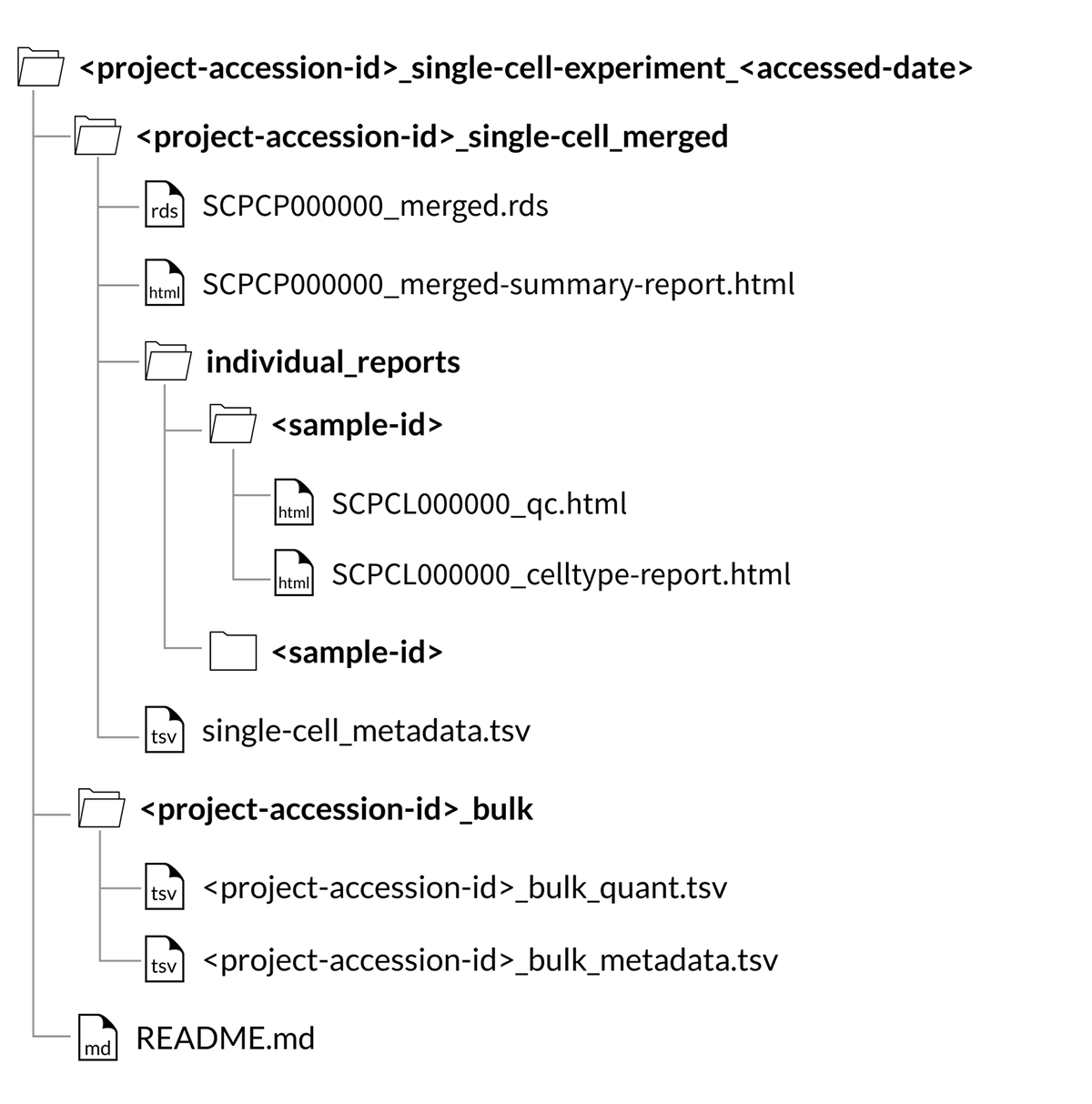
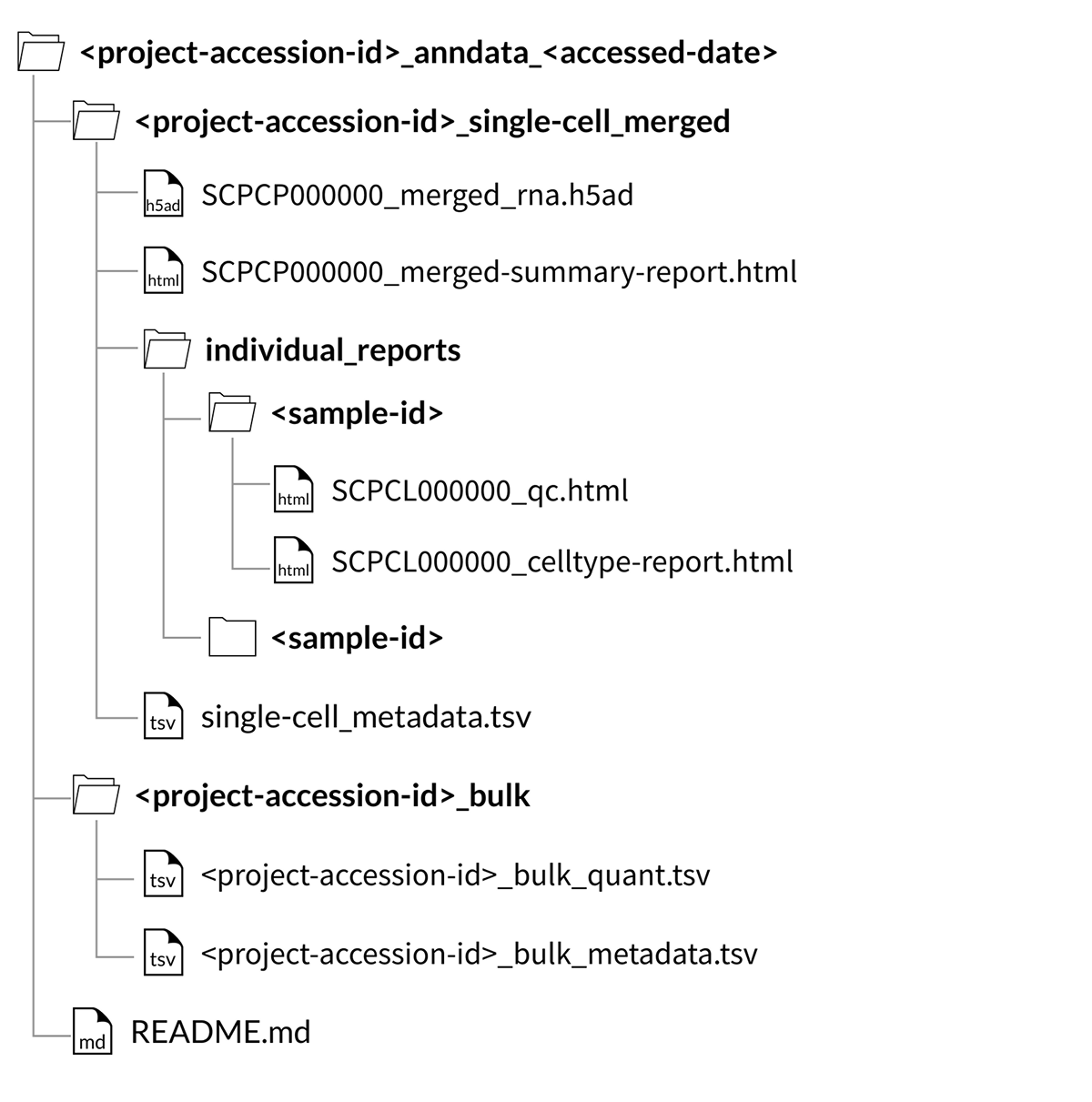
Merged object downloads
Merged object downloads contain all single-cell or single-nuclei gene expression data for a given ScPCA project within a single object, provided as either a SingleCellExperiment object (.rds file) or an AnnData object (.h5ad file).
The object file, SCPCP000000_merged.rds or SCPCP000000_merged_rna.h5ad, contains both a raw and normalized counts matrix, each with combined counts for all samples in an ScPCA project.
In addition to the counts matrices, the SingleCellExperiment or AnnData object stored in the file includes the results of library-weighted dimensionality reduction using both principal component analysis (PCA) and UMAP.
See the section on merged object processing for more information about how merged objects were created.
If downloading a project that contains at least one CITE-seq library, the quantified CITE-seq expression data will also be merged.
In SingleCellExperiment objects (.rds files), the CITE-seq expression data is provided as an alternative experiment in the same object as the gene expression data.
However, for AnnData objects, (.h5ad files), the quantified CITE-seq expression is instead provided as a separate file called SCPCP000000_merged_adt.h5ad.
For any projects containing bulk RNA-seq data, a separate folder SCPCP000000_bulk containing two tab-separated value files, SCPCP000000_bulk_quant.tsv and SCPCP000000_bulk_metadata.tsv, will also be included in the project download.
See the section on bulk RNA-seq for more information.
Every download also includes a single single-cell_metadata.tsv file containing metadata for all libraries included in the merged object.
For a full description of this file’s contents, refer to the metadata section below.
Every download includes a summary report, SCPCP000000_merged-summary-report.html, which provides a brief summary of the samples and libraries included in the merged object.
This includes a summary of the types of libraries (e.g., single-cell, single-nuclei, with CITE-seq) and sample diagnoses included in the object, as well as UMAP visualizations highlighting each library.
Every download also includes the individual QC report and, if applicable, cell type annotation reports for each library included in the merged object.
Download folder structure for SingleCellExperiment merged downloads:
Download folder structure for AnnData merged downloads:
Download folder structure for AnnData merged downloads with CITE-seq (ADT) data:
Custom datasets
You can create a custom dataset with any combination of individual samples and projects with your choice of modalities and data format.
Custom datasets are referred to as My Dataset within the portal.
The Add to Dataset button allows you to add projects and selected samples to My Dataset.
You can select the data formats and modalities for each project or sample before you add it to My Dataset.
The My Dataset button on the top right of the portal can then be used to view and download the custom dataset as a single zip file.
Each zip file will be named with a unique dataset ID, the chosen data format (either single-cell-experiment or anndata), and the date you accessed the data on the ScPCA Portal.
Note that a custom dataset can only contain single-cell data in one data format, SingleCellExperiment objects (.rds files) or AnnData objects (.h5ad files) (see FAQ for more information).
If a sample has spatial transcriptomics data, you can check the Spatial modality box to include the spatial transcriptomics data in My Dataset.
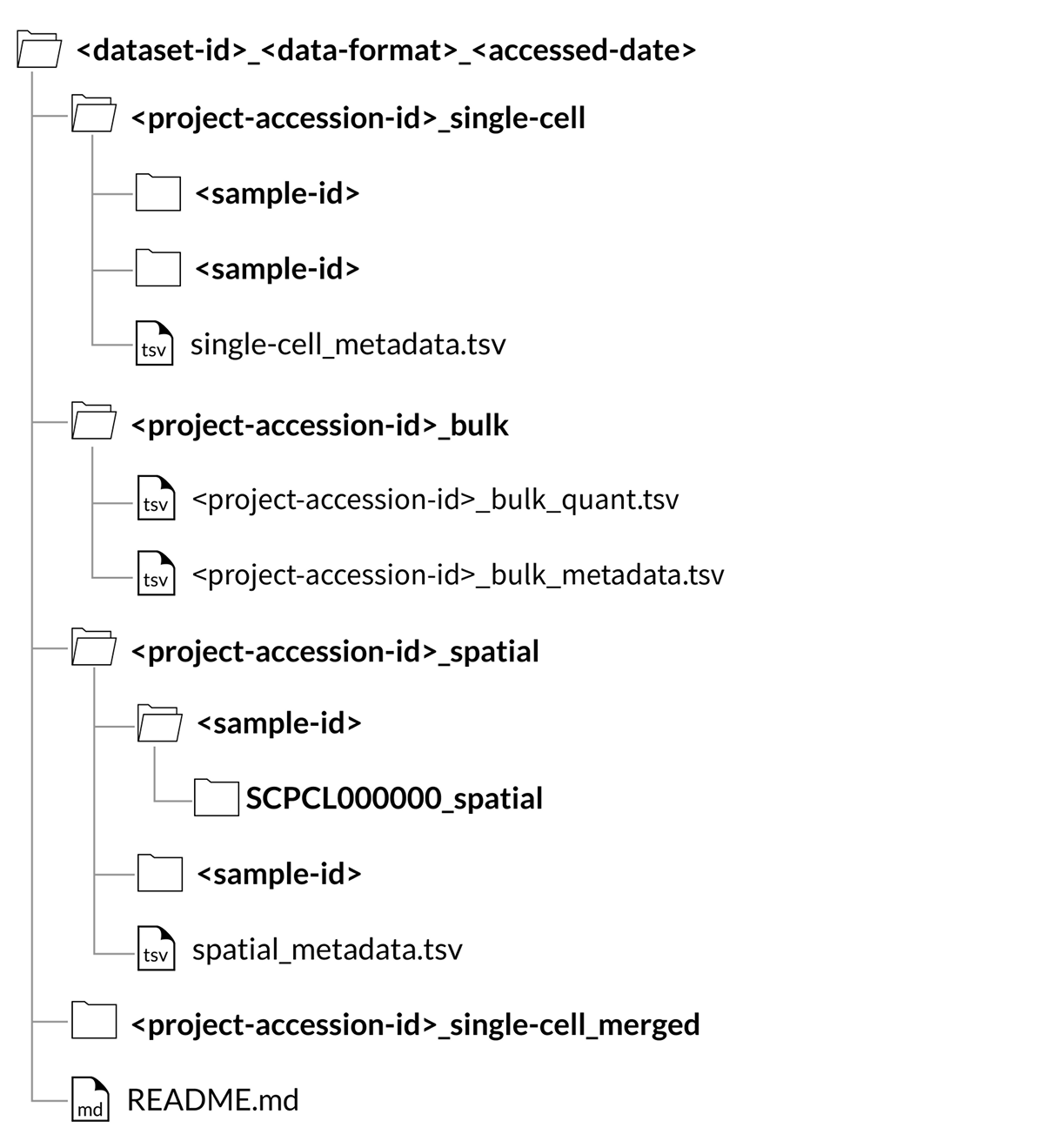
Data for all samples included in My Dataset will be organized in folders labeled with the unique project identifier and modality, where each folder contains data for all samples from a single project with the same modality (either single-cell, spatial, or bulk).
Each project folder will also contain an appropriate metadata file, either single-cell_metadata.tsv (single-cell), spatial_metadata.tsv (spatial), or SCPCP000000_bulk_metadata.tsv (bulk).
For more information on available data formats and modalities, see the section describing download options.
If downloading a project as a merged object, the project folder will contain a _merged suffix.
If any samples included in My Dataset contain associated CITE-seq data, the quantified CITE-seq expression data will be included when downloading single-cell expression data.
For SingleCellExperiment objects (.rds files), the quantified CITE-seq expression is included in the same file as the gene expression data.
For AnnData objects (.h5ad files), the quantified CITE-seq expression data is included as a separate file with the suffix _adt.h5ad.
The below image shows the expected file structure for an example custom dataset. For more details about the project folder contents for each data format and modality, see the Project downloads section.
Download folder structure for custom downloads:
Portal-wide Downloads
The Portal-wide Download page can be used to download all metadata or gene expression data for all samples on the Portal at once.
All single-cell and single-nuclei gene expression data from the Portal can be downloaded as a single zip file containing data stored as either SingleCellExperiment objects (.rds files) or AnnData objects (.h5ad files).
This zip file includes data for any multiplexed samples.
All spatial data for any samples sequenced using spatial transcriptomics are available separately as a zip file.
When downloading any of the available Portal-wide data downloads, all relevant metadata and bulk RNA-seq data is also included.
Each zip file will be named with the chosen data format (single-cell-experiment, anndata, or spaceranger) and the date you accessed the data on the ScPCA Portal.
Each zip file will contain a folder for each project with gene expression data for all samples in that project as either individual objects or a single merged object, depending on your selection.
For any projects containing bulk RNA-seq data, a separate folder SCPCP000000_bulk containing two tab-separated value files, SCPCP000000_bulk_quant.tsv and SCPCP000000_bulk_metadata.tsv, will also be included.
See the section on bulk RNA-seq for more information.
As with individual project and custom datasets, the quantified CITE-seq expression data will be included when downloading single-cell expression data.
For SingleCellExperiment (R) downloads, the quantified CITE-seq expression is included in the same file as the gene expression data.
For AnnData (Python) downloads, the quantified CITE-seq expression data is included as a separate file with the suffix _adt.h5ad.
SingleCellExperiment Portal-wide download structure

AnnData Portal-wide download structure

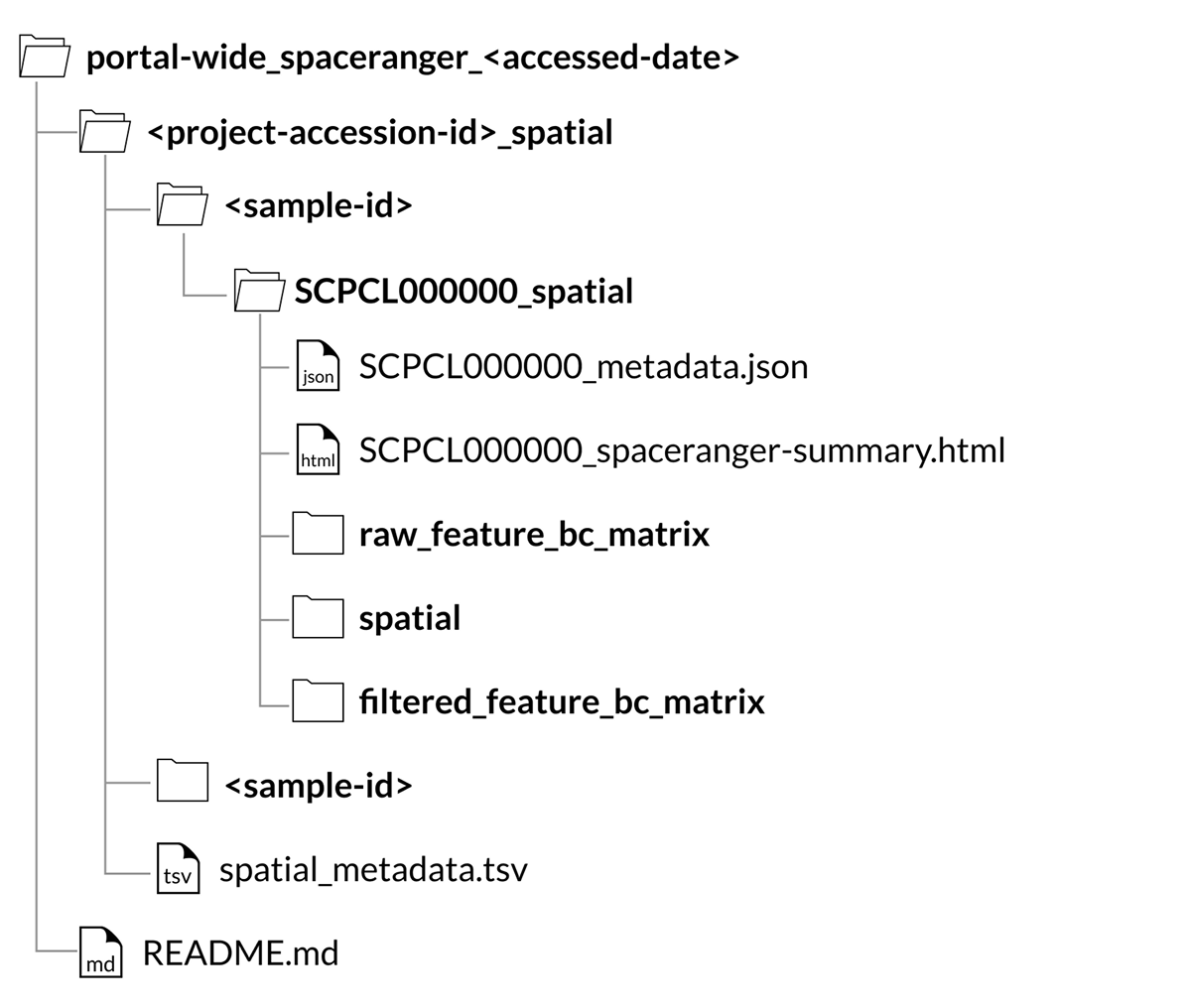
Spatial Portal-wide download structure
Portal-wide downloads as merged objects
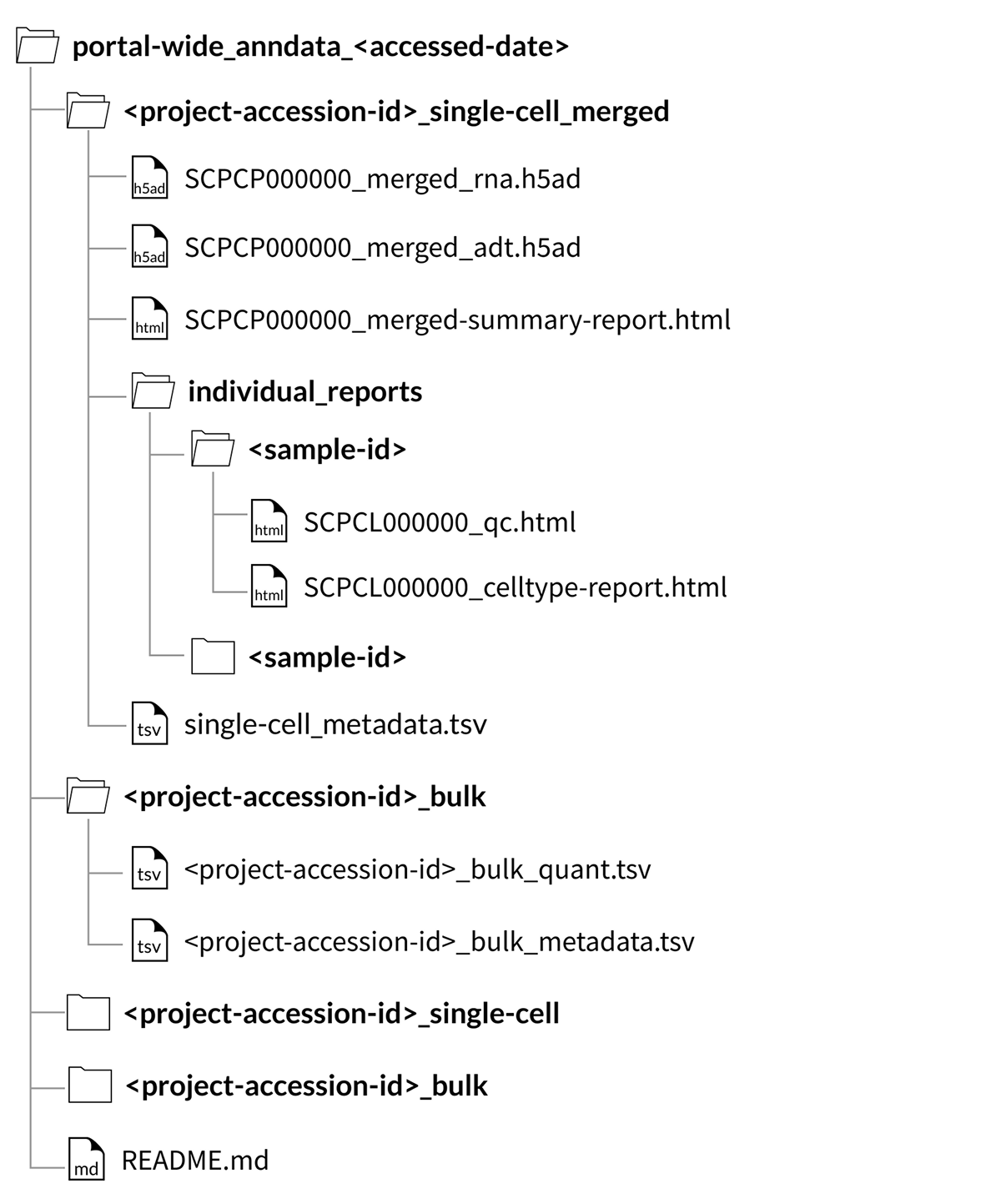
You can choose to download all single-cell and single-nuclei samples from the Portal as merged objects for each project by checking “Merge samples into one object per project”.
Merged objects contain gene expression for all samples in a given project in a single file.
This download includes a folder for each project that contains a single merged object (SCPCP000000_merged.rds or SCPCP000000_merged.h5ad), a merged summary report (SCPCP000000_merged-summary-report.html), a single metadata file (single-cell_metadata.tsv), and all individual QC reports and, if applicable, cell type annotation reports for each library included in the merged object for that project.
Note that downloading all data using this option will not download a merged object with all samples from all projects, but a single merged object for each project.
Portal-wide download structure for merged SingleCellExperiment objects

Portal-wide download structure for merged AnnData objects
Metadata-only downloads
The Portal-wide metadata download is a single TSV file containing the metadata for all samples with associated single-cell RNA-seq, single-nuclei RNA-seq, or spatial transcriptomics data available on the Portal. A table describing all columns included in the file can be found in the metadata section below.
Gene expression data
Single-cell or single-nuclei gene expression data is provided as either SingleCellExperiment objects (.rds files) or AnnData objects (.h5ad files).
Three files will be provided for each library included in the download - an unfiltered counts file, a filtered counts file, and a processed counts file.
The unfiltered counts file, SCPCL000000_unfiltered.rds or SCPCL000000_unfiltered_rna.h5ad, contains the counts matrix, where the rows correspond to genes or features and the columns correspond to cell barcodes.
Here, all potential cell barcodes that are identified after running alevin-fry are included in the counts matrix.
The object also includes summary statistics for each cell barcode and gene, as well as metadata about that particular library, such as the reference index and software versions used for mapping and quantification.
The filtered counts file, SCPCL000000_filtered.rds or SCPCL000000_filtered_rna.h5ad contains a counts matrix with the same structure as above.
The cells in this file are those that remain after filtering using emptyDrops.
As a result, this file only contains cell barcodes that are likely to correspond to true cells.
The processed counts file, SCPCL000000_processed.rds or SCPCL000000_processed_rna.h5ad, contains both the raw and normalized counts matrices.
The filtered counts file is further filtered to remove low quality cells, such as those with a low number of genes detected or high mitochondrial content.
This file contains the raw and normalized counts data for cell barcodes that have passed both levels of filtering.
In addition to the counts matrices, the SingleCellExperiment or AnnData object stored in the file includes the results of dimensionality reduction using both principal component analysis (PCA) and UMAP.
See Single-cell gene expression file contents for more information about the contents of the SingleCellExperiment and AnnData objects and the included statistics and metadata.
See also Getting started with an ScPCA dataset.
QC report
The included QC report, SCPCL000000_qc.html, serves as a general overview of each library, including processing information, summary statistics and general visualizations of cell metrics.
Cell type report
The cell type report, SCPCL000000_celltype-report.html, includes an overview of cell type annotations present in the processed objects.
This report contains details on methodologies used for cell type annotation, information about reference sources, comparisons among cell type annotation methods, and diagnostic plots.
For more information on how cell types were annotated, see the section on Cell type annotation.
If the downloaded library was from a cell line sample, no cell type annotation will have been performed. Therefore, there will be no cell type report in the download for these libraries.
Metadata
Included with each download is a single-cell_metadata.tsv file containing relevant metadata for each sample included in the download.
Each row corresponds to a unique sample/library combination and contains the following columns:
column_id |
contents |
|---|---|
|
Sample ID in the form |
|
Library ID in the form |
|
Tumor type |
|
Subcategory of diagnosis or mutation status (if applicable) |
|
At what stage of disease the sample was obtained, either diagnosis or recurrence |
|
Age provided by submitter |
|
Whether age is the age at diagnosis ( |
|
Sex of patient that the sample was obtained from |
|
Where in the body the tumor sample was located |
|
Unique id corresponding to the donor from which the sample was obtained |
|
Original sample identifier from submitter |
|
Submitter name/id |
|
The organism the sample was obtained from (e.g., |
|
|
|
|
|
NCBI taxonomy term for organism, e.g. |
|
For Homo sapiens samples, a |
|
|
|
|
|
|
|
10x kit used to process library |
|
Total number of reads processed by |
|
Number of reads successfully mapped |
|
Total number of cells detected by |
|
Number of cells after filtering with |
|
Number of cells after filtering with |
|
Number of cells after removing low quality cells |
|
Boolean indicating if the library has associated cell hashing data |
|
Boolean indicating if |
|
Boolean indicating whether or not the sample was obtained from a cell line |
|
Boolean indicating if the library contains multiplexed samples |
|
Boolean indicating whether or not the sample was obtained from a patient-derived xenograft |
|
Project ID in the form |
|
Name of primary investigator |
|
Title of project |
|
Ensembl version of genome used for mapping with |
|
Name of index used for mapping with |
|
Version of 10x Genomics’ Space Ranger used for mapping, only present for spatial transcriptomics samples |
|
Version of |
|
Version of |
|
Types of counts matrices included in |
|
The method used for cell filtering. One of |
|
Method used by the Data Lab to filter low quality cells prior to normalization. Either |
|
The minimum cutoff for the probability of a cell being compromised, as calculated by |
|
The minimum cutoff for the number of unique genes detected per cell |
|
The method used for normalization of raw RNA counts. Either |
|
Methods used to calculate demultiplexed sample numbers. Only present for multiplexed libraries |
|
Samples included in multiplexed library. Only present for multiplexed libraries |
|
Date sample was processed through |
|
The URL to the |
|
Version of |
|
Commit hash of |
Project-specific metadata will contain all columns listed in the table above and any additional project-specific columns, such as treatment or outcome.
Metadata pertaining to processing will be available in this table and inside of the SingleCellExperiment and AnnData objects.
See the SingleCellExperiment experiment metadata section for more information on metadata columns that can be found in the SingleCellExperiment object.
See the AnnData experiment metadata section for more information on metadata columns that can be found in the AnnData object.
For projects with bulk RNA-seq data, a bulk metadata file (e.g., SCPCP000000_bulk_metadata.tsv) will be included for project downloads.
This file will contain fields equivalent to those found in the single-cell_metadata.tsv related to processing the sample, but will not contain patient or disease specific metadata (e.g. age, sex, diagnosis, subdiagnosis, tissue_location, or disease_timing).
Multiplexed sample libraries
For libraries where multiple biological samples were combined via cellhashing or similar technology (see the FAQ section about multiplexed samples), the organization of the downloaded files and metadata is slightly different.
Note that multiplexed sample libraries are only available as SingleCellExperiment objects, and are not currently available as AnnData objects.
When downloading an entire project, the counts and QC files will be organized by the set of samples that comprise each library, rather than in individual sample folders.
These sample set folders are named with an underscore-separated list of the sample ids for the libraries within, e.g., SCPCS999990_SCPCS999991_SCPCS999992.
Bulk RNA-seq data, if present, will follow the same format as bulk RNA-seq for single-sample libraries.
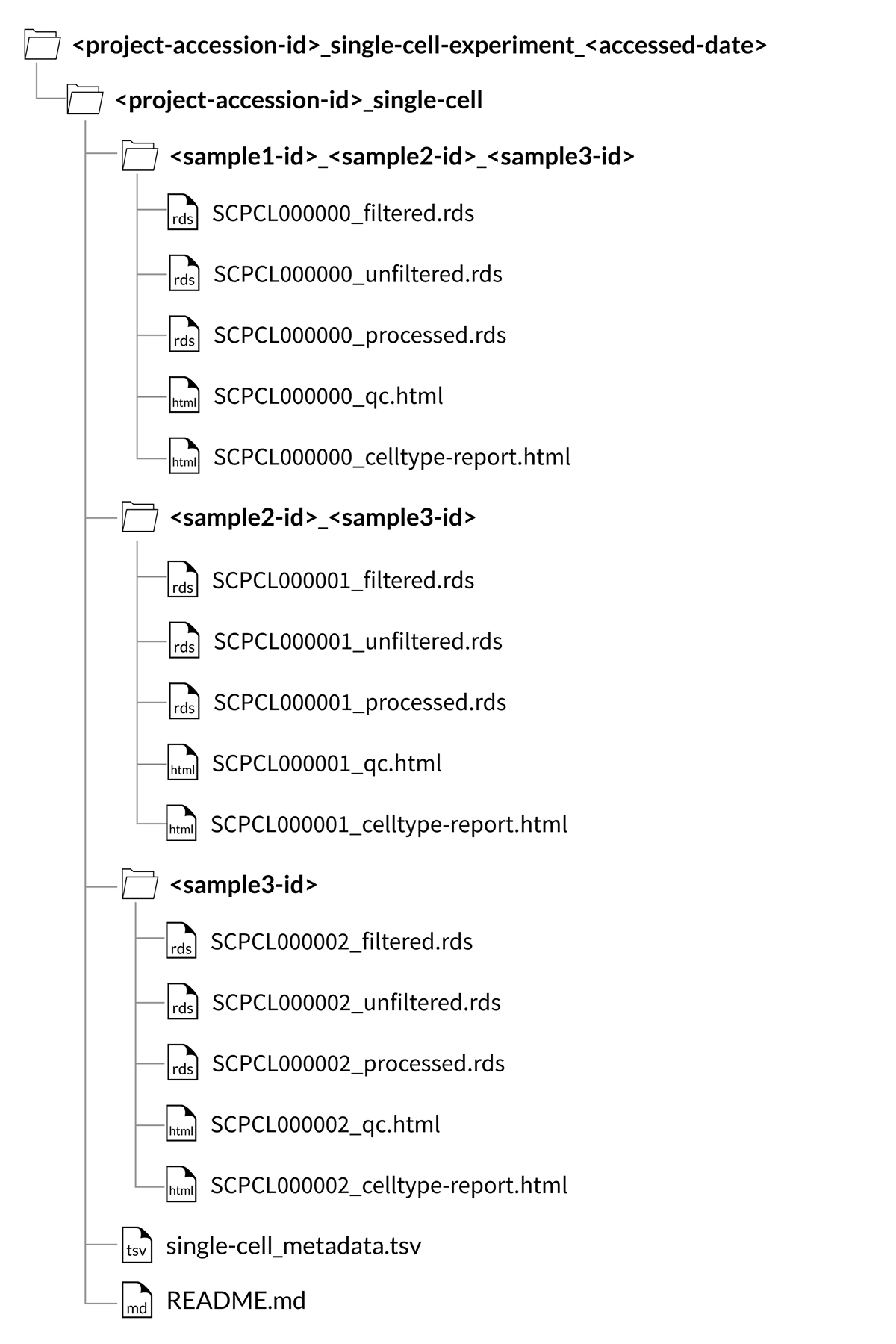
Because we do not perform demultiplexing to separate cells from multiplexed libraries into sample-specific count matrices, sample downloads from a project with multiplexed data will include all libraries that contain the sample of interest, but these libraries will still contain cells from other samples.
For more on the specific contents of multiplexed library SingleCellExperiment objects, see the Additional SingleCellExperiment components for multiplexed libraries section.
The metadata file for multiplexed libraries (single-cell_metadata.tsv) will have the same format as for individual samples, but each row will represent a particular sample/library pair, meaning that there may be multiple rows for each scpca_library_id, one for each scpca_sample_id within that library.
In addition, the demux_cell_count_estimate column will contain an estimate of the number of cells from the sample in the library (after demultiplexing) in the sample/library pair.
Spatial transcriptomics libraries
If a sample includes a library processed using spatial transcriptomics, you can obtain the spatial transcriptomics output files by selecting Spatial as the modality (see more on modality download options).
If downloading an entire project using the Download Now button, you will need to download the spatial data separately from the single-cell and single-nuclei gene expression data.
If creating and downloading a custom dataset by using the Add to Dataset button, you will be able to select both Single-cell and Spatial to be included in the download.
Alternatively, you can download all of the spatial transcriptomic data from the Portal on the Portal-wide Downloads page.
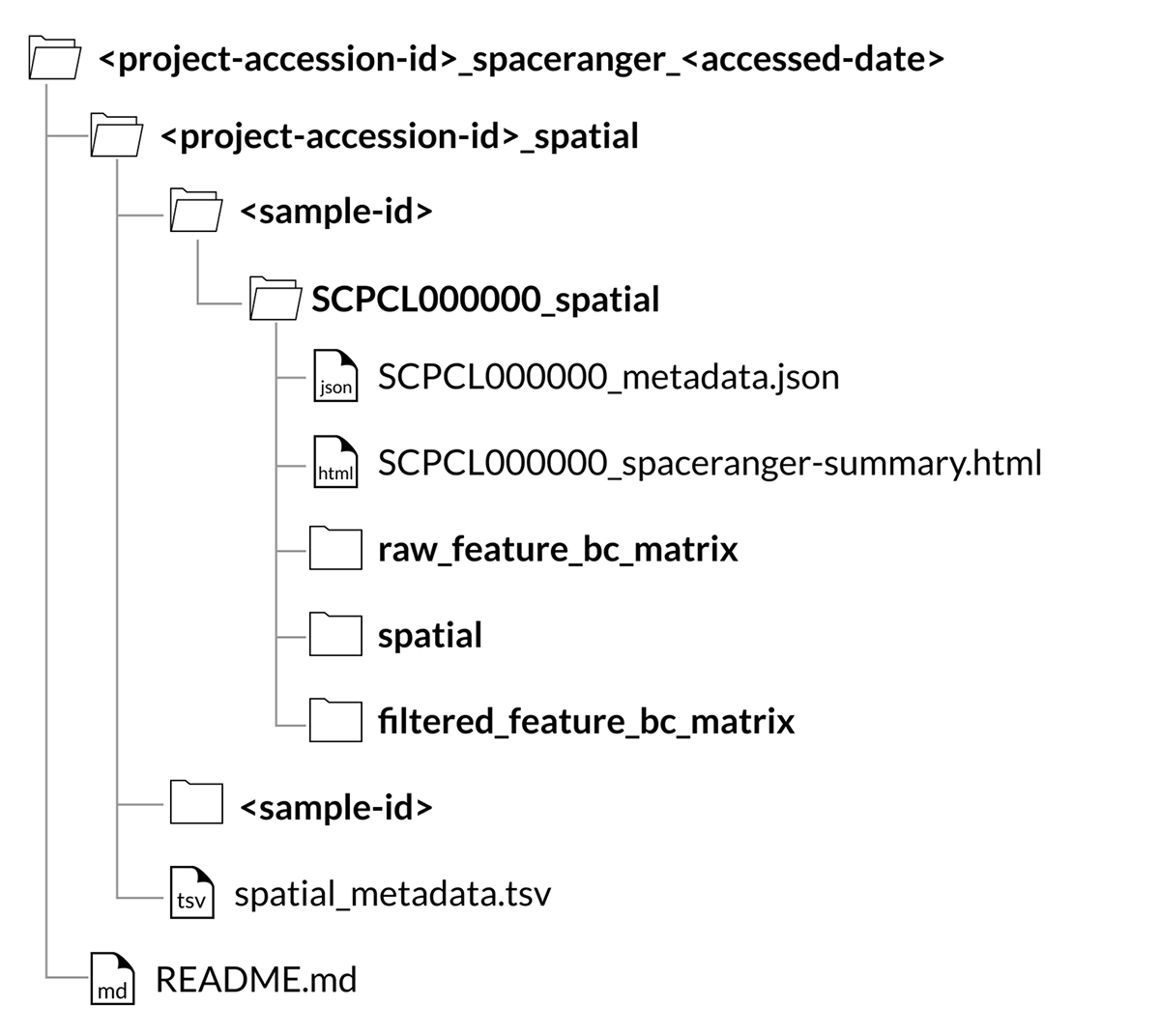
For all spatial transcriptomics libraries, a SCPCL000000_spatial folder will be nested inside the corresponding sample folder in the download.
Inside that folder will be the following folders and files:
A
raw_feature_bc_matrixfolder containing the unfiltered counts filesA
filtered_feature_bc_matrixfolder containing the filtered counts filesA
spatialfolder containing images and position informationA
SCPCL000000_spaceranger-summary.htmlfile containing the summary html report provided by Space RangerA
SCPCL000000_metadata.jsonfile containing library processing information.
A full description of all files included in the download for spatial transcriptomics libraries can also be found in the spaceranger count documentation.
Every download also includes a single spatial_metadata.tsv file containing metadata for all libraries included in the download.
Bulk RNA-seq
Some projects include samples that were sequenced using bulk RNA-seq alongside any single-cell and/or single-nucleus RNA-seq. For more details on including bulk RNA-seq in your download, see the documentation on download options.
A separate folder labeled with the project accession ID and _bulk suffix will be included for each project containing bulk RNA-seq in the download.
This folder contains two tab-separated value files, SCPCP000000_bulk_quant.tsv and SCPCP000000_bulk_metadata.tsv.
The SCPCP000000_bulk_quant.tsv file contains a gene by sample matrix (each row a gene, each column a sample) containing raw gene expression counts quantified by salmon.
The SCPCP000000_bulk_metadata.tsv file contains associated metadata for all samples with bulk RNA-seq data.
This file will contain fields equivalent to those found in the single-cell_metadata.tsv related to processing the sample, but will not contain patient or disease specific metadata (e.g. age, sex, diagnosis, subdiagnosis, tissue_location, or disease_timing).
See also processing bulk RNA samples.
Programmatic downloads from the ScPCA Portal
We provide an R package, ScPCAr, to facilitate programmatic access to the ScPCA Portal.
This package allows you to search for and download data from the ScPCA Portal directly within R.
An example of basic usage of the ScPCAr package follows:
library(ScPCAr)
# First, look at the terms of use
view_terms()
# Get an authentication token for use with the ScPCA Portal
auth_token <- get_auth(email = "your.email@example.com", agree = TRUE)
# Get the sample metadata for a project
sample_metadata <- get_sample_metadata(project_id = "SCPCP000001")
# Download data for a sample
# this function returns a vector of the downloaded file paths
file_paths <- download_sample(
sample_id = "SCPCS000001",
auth_token = auth_token,
destination = "scpca_data",
format = "sce"
)
# select and read in the processed SingleCellExperiment object
processed_data <- grep("_processed.rds$", file_paths)
sce <- readRDS(processed_data)
Please see the package documentation for more details about installation and usage. Source code for the package can be found on GitHub. Information about working with downloaded files can be found in our Getting started with an ScPCA dataset guide.